
Dott.ssa Mercedes Pineda, Servicios de Neuropediatria, Bioquimica y Endocrinologia, Hospital Sant Joan de Deu, Clinic, Barcelona, Spain
Studio effettuato presso l’Ospedale Sant Joan de Deu di Barcellona, su un campione di 40 pazienti di età compresa tra 19 mesi e 28 anni, con lo scopo di:
- studiare la relazione tra fenotipo e genotipo (MECP2), con particolare riferimento al tipo di crisi epilettica, al pattern video-EEG e alla risposta ai farmaci antiepilettici;
- trovare il farmaco antiepilettico più efficace (in monoterapia o in associazione);
- studiare il fenomeno delle crisi “autoindotte” (la paziente è in grado di provocare crisi riflesse attraverso l’iperventilazione o la pressione ritmica delle mani), che è infrequente nei classici pazienti epilettici;
- valutare l’influenza di un buon controllo dell’epilessia sulla qualità di vita.
TIPI DI EPILESSIA
- crisi generalizzate,
- assenze,
- spasmi,
- crisi focali,
- crisi riflesse,
- stato confusionale,
- stato di male epilettico in sonno.
EPILESSIA RIFLESSA
Si tratta di crisi provocate da stimoli precipitanti come stimoli visivi, reazioni di allarme, alimentazione e musica, acqua calda e altri stimoli somatosensitivi e propriocettivi.
Dall’analisi dei risultati ottenuti dal follow-up è emerso che le crisi sono state maggiormente eterogenee nel gruppo di pazienti in cui non è stata identificata la mutazione genica e nelle forme meno frequenti di mutazione.
La diagnosi genetica fornisce indicazioni sulla probabilità di comparsa di epilessia, sull’età d’esordio delle crisi, ma non sul tipo di crisi e sulla risposta al trattamento.
Nel campione seguito si è dimostrata una prevalenza di epilessia del 75%.
Nel 16% dei pazienti è stato evidenziato uno stato di male epilettico durante il follow-up.
Le forme più comuni osservate sono quelle generalizzate tonico-cloniche, mentre tra le forme focali hanno maggiore incidenza le forme toniche motorie.
I pattern video-EEG predominanti sono i parossismi multifocali, attivati in sonno.
TERAPIA ANTIEPILETTICA
La scelta dei farmaci antiepilettici dipende dal tipo di crisi e dalle alterazioni riscontrate alla registrazione video-EEG. Nella maggior parte dei casi sono utilizzati Valproato e Carbamazepina, farmaci con i quali sono stati ottenuti i migliori risultati, sia in mono- che in politerapia.
In 3 pazienti è stato possibile sospendere il trattamento antiepilettico, senza la ricomparsa di nuove crisi.
CDKL5 NELLA VARIANTE ATIPICA DELLA SINDROME DI RETT CON EPILESSIA PRECOCE IN PAZIENTI SPAGNOLI
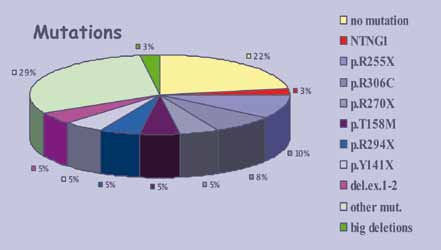
Nella forma classica della Sindrome di Rett si riscontrano mutazioni in MECP2 nell’80% dei casi, mentre nelle varianti tali mutazioni si osservano solo nel 20-40% dei pazienti.
CDKL5 è un gene situato sul cromosoma X (Xp22), che codifica per una proteina chinasi ciclina dipendente che modula l’espressione di MECP2 (con cui condivide vie metaboliche comuni di interregolazione e un fenotipo simile) ed è coinvolta nella maturazione e migrazione neuronale.
Presso l’Ospedale Sant Joan de Deu de Barcelona sono stati esaminati 383 pazienti con Sindrome di Rett e 5 pazienti di sesso maschile con encefalopatia epilettica farmacoresistente.
In 123 pazienti con Sindrome di Rett è stata esclusa la presenza di mutazioni a carico di MECP2.
28 di questi pazienti presentano epilessia:
- 20 pazienti risultano affetti dalla forma classica di Sindrome di Rett;
- 4 presentano epilessia precoce;
- 2 presentano forma congenita;
- 2 pazienti presentano forma a regressione tardiva.
Questi 28 pazienti e i 5 pazienti maschi con encefalopatia epilettica farmacoresistente sono stati sottoposti a studio genetico per la ricerca di mutazioni a carico del gene CDKL5.
Non sono state identificate mutazioni del gene nei 5 maschi affetti da encefalopatia epilettica farmacoresistente, mentre mutazioni di CDKL5 sono state identificate in 11 pazienti con forma atipica di Rett, nella variante con epilessia farmacoresistente.
TRIAL TERAPEUTICO PER I DISTURBI COMPORTAMENTALI NELLA SINDROME DI RETT
Sono stati studiati due farmaci, utilizzati nel tentativo di migliorare l’umore e il comportamento:
- Venlafaxina (inibitore selettivo del reuptake della Serotonina);
- Citalopram (inibitore selettivo del reuptake di Serotonina e Noradrenalina).
L’ipotesi su cui si basa la scelta di questi farmaci è che alterati livelli di catecolamine a livello cerebrale siano associati ad un’anomala funzione sinaptica, responsabile di cambiamenti dell’umore e del comportamento nelle pazienti affette da Sindrome di Rett.
I comportamenti possono essere quindi modificati con farmaci che aumentano le concentrazioni di Serotonina e Noradrenalina a livello sinaptico.
Lo studio è stato effettuato su bambine con problemi comportamentali significativi che, per intensità e frequenza, interferiscono con la qualità di vita loro e della famiglia e in bambine che presentano mutazione del gene MECP2.
La valutazione dell’efficacia dei farmaci somministrati è basata sulla valutazione clinica e sull’utilizzo di questionari sulla qualità di vita, associati ad esami specifici come EEG, ECG, esami ematici di routine, dosaggio vit. D e IGF-1.
Dopo l’avvio della terapia, ogni paziente è stata monitorata telefonicamente, ogni settimana nel primo mese, poi con scadenza mensile nei restanti 2 mesi.
