
Dott.ssa Silvia Russo, Francesca Cogliati
Istituto Auxoligico Milano
Accanto alla forma classica della sindrome di Rett si riconoscono delle forme definite “atipiche”, che differiscono dalla forma classica per la severità e il tipo di epilessia, per la presenza di linguaggio seppure ridotto per l’età di esordio della regressione, che può essere più tardiva oppure ci possono essere forme congenite (in cui il deficit cognitivo è presente sin dalla nascita). Solo il 40% delle pazienti con una forma atipica di Rett ha mutazione nel gene principale MECP2. Si distinguono quindi a) varianti parlanti ed in questa coorte si trovano bambine con mutazioni nel gene principale, b) varianti ad insorgenza precoce dell’epilessia detta anche di Hanefeld con mutazioni nel gene CDKL5 e c) forme congenite con mutazioni nel gene FOXG1.
La variante con insorgenza precoce dell’epilessia ed il gene CDKL5 rappresenta la seconda più importante forma della sindrome di Rett. Dal punto di vista clinico condivide con la forma classica un grave ritardo nello sviluppo psicomotorio, aprassia manuale e la presenza di stereotipie della mani, ma si distingue dalla prima per l’assenza di un vero e proprio periodo perinatale normale e soprattutto dall’insorgenza, fra le prime settimane di vita e i 5 mesi, di epilessia, che può manifestarsi con spasmi infantili e crisi resistenti al trattamento farmacologico. Circa il 9% delle bambine che manifesta insorgenza precoce di epilessia ed il 28% epilessia ad insorgenza precoce e spasmi infantili presentano mutazioni in CDKL5 (Nemos et al, 2009). L’epilessia è tonica refrattaria o mioclonica nel 75% dei casi, mentre si ha remissione spontanea nel 25% dei casi.
A differenza della forma classica, le bimbe con la variante Hanefeld presentano una circonferenza cranica generalmente nella norma, una grave ipotonia e spesso non hanno le disfunzioni del sistema neurovegetativo tipiche della forma classica, né mantengono l’intenso contatto oculare che contraddistingue la sindrome. Non sono riportati casi di femmine portatrici sane, né casi familiari.
GENE E NUOVE ISOFORME
Nel 2005 sono state identificate le prime mutazioni nel gene CDKL5 (Cyclin-Dependent kinase-like 5), che rappresenta quindi il secondo gene causativo della sindrome. Tale gene è localizzato in Xp22 e presenta alcune isoforme: la isoforma I, contenente l’esone 1, che consta di 21 frammenti, detti ‘esoni’, che è espressa in un’ampia varietà di tessuti e la isoforma II, contenente gli esoni 1a e 1b, che consta di 22 esoni ed è trascritta solo nei testicoli e, a livelli molto bassi, nel cervello fetale. Negli ultimi 2 anni sono state identificate 2 nuove isoforme: a) isoforma ‘16b’, che contiene un esone in più localizzato fra il vecchio esone 16 e il 17, il cui trascritto si trova in grande quantità nei fibroblasti umani e nel cervello di topo, e b) isoforma più corta con soli 18 esoni, in cui l’esone 18 è più lungo poiché continua per 170 nucleotidi nella porzione che nelle altre isoforme è considerata parte intronica, non codificante. Tale forma è ubiquitariamente espressa. L’identificazione di nuove isoforme riveste importanza a fini diagnostici, poiché per avere un’analisi esaustiva, è necessario studiare anche tali regioni che prima non venivano indagate. Tuttavia uno studio dell’esone 16b eseguito in tre numerosi gruppi di pazienti con Rett classica o reminiscente (Fichou 2011), con RTT classica o Hanefeld (Rademacher 2011), con rett atipica, ritardo mentale e autismo (White 2010), non hanno evidenziato alcuna alterazione, suggerendo che mutazioni patogenetiche in queste nuove regioni siano rare.
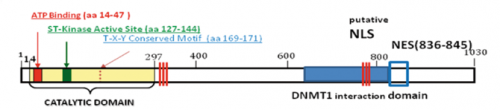
FUNZIONE DEL GENE
Dal punto di vista funzionale CDKL5 codifica per una proteina con attività chinasica, cioè in grado di fosforilare altre proteine, oltre che se stessa; la fosforilazione è una funzione importante perché rappresenta uno dei segnali di comunicazione fra le proteine all’interno della cellula, che ne favorisce l’attivazione/disattivazione e comunque la variazione della loro funzionalità. La regione preposta a tale importante funzione si chiama dominio catalitico, è codificato dai primi 11 esoni del gene, e contiene 3 siti principali: quello per il legame all’ATP (la molecola che fornisce la energia necessaria perché avvengano tali reazioni di fosforilazione), il sito attivo treonin-chinasico (ove si lega la regione di un’altra proteina che viene fosforilata da CDKL5) e un sito di fosforilazione Thr-Glu-Tyr, cioè una regione consenso dove la proteina CDKL5 può essere fosforilata da altre proteine o da sé medesima.
Il gene CDKL5 viene espresso attivamente nel primo periodo postnatale e, a differenza di MECP2, nel cervello adulto si trova solo nei neuroni, ma non nelle cellule della astroglia, è presente sia nel citoplasma delle cellule in divisione, sia nel nucleo e si trasferisce da un compartimento all’altro, in maniera differente a seconda dell’area del cervello e del momento dello sviluppo. (Rusconi, 2008). Nel citoplasma CDKL5 è coinvolto nel rimodellamento actinico e nella morfogenesi neuronale, cioè nella crescita dell’assone (diramazione che dirige l’impulso nervoso in direzione centrifuga e termina con le sinapsi), e nella arborizzazione dendritica, cioè delle fibre minori che si ramificano a partire dal neurone e che trasportano il segnale nervoso verso il corpo cellulare del neurone. A livello del nucleo sono state proposte diverse funzioni per la proteina CDKL5: a) studi in vitro, hanno dimostrato che CDKL5 interagisce con MECP2 (Mari, 2005) e con la DNA metiltransferasi 1 (DNMT1) (Kameshita, 2008), suggerendo che possa regolare la metilazione del DNA e il legame di MECP2 stesso al DNA, b) studi in vivo (Carouge, 2010) in neuroni di ratto dimostrano che MECP2 agisce su di CDKL5 reprimendolo. Infatti CDKL5 viene inibito da MECP2 attraverso il legame di quest’ultimo ad una regione ricca di citosine metilate che copre il promotore. Tale scoperta ribadisce il coinvolgimento in un medesimo meccanismo di azione delle due maggiori proteine responsabili della sindrome di Rett; inoltre c) si è dimostrato che in vitro CDKL5 a sua volta fosforila MECP2; quindi il legame fra le due proteine e le due forme della sindrome è duplice: se da un lato le mutazioni in CDKL5 possono alterare la fosforilazione di MECP2, portando nelle bambine con variante Hanefeld, ad un’attività ridotta di MECP2 che genera una serie di sintomi della sindrome di Rett, ci si aspetterebbe viceversa che mutazioni in MECP2 nelle pazienti con Rett classica, che riducono la sua attività di legame alle citosine metilate nella regione a monte di CDKL5, determinino un incremento della espressione di CDKL5, il cui significato sul fenotipo sarebbe interessante da valutare. Infine d) CDKL5 a livello nucleare sembrerebbe coinvolto in una nuova funzione, poiché co-localizza a livello di regioni specifiche dette ‘nuclear speckles’ dove si trovano i fattori che regolano il ‘taglio’ (splicing) dell’RNA, molecola che rappresenta la fase di transizione dal gene alla proteina. Sia l’over-espressione che un abbassamento della quantità di CDKL5 portano ad una alterazione di tali strutture, mediata probabilmente dalla sua attività di fosforilazione sulle proteine che fanno parte dei ‘nuclear speckles’ (Broccoli).
ANALISI MUTAZIONALE
La sensibilità clinica del test di screening per CDKL5, cioè la possibilità di identificare pazienti positive fra le pazienti di sesso femminile con caratteristiche Rett-like ma negative all’analisi per MECP2 è riportata pari al 7,8%, valore che sale al 14,3% se come criterio diagnostico si aggiunge l’insorgenza di epilessia precoce e farmaco resistente prima dei tre mesi di età (Intusoma, 2011), che rappresenta quindi il tratto fenotipico principale in grado di indirizzare il clinico a suggerire l’analisi per la paziente.
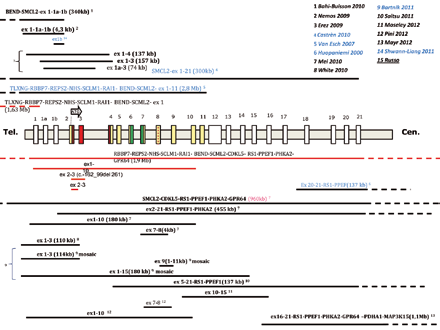
Ad oggi sono state identificate in letteratura circa 80 pazienti con mutazione in CDKL5, fra cui anche 13 maschi. La maggior parte delle mutazioni, circa 50, sono puntiformi, cioè interessano uno o pochi nucleotidi della sequenza e comunque sono evidenziabili tramite la tecnica del sequenziamento. Si tratta di mutazioni missenso all’interno del dominio catalitico, mutazioni non senso causanti la terminazione prematura della proteina, distribuite lungo l’intera sequenza del gene, varianti di splicing e mutazioni frameshifts. Le rimanenti 30 interessano delezione di regioni più estese, comprendenti uno o più esoni del gene, e non possono essere evidenziate con la tecnica precedente, ma con la tecnica dell’MLPA o della PCR quantitativa (Real Time).
Nel laboratorio dell’IAI, dal 2005, è stato analizzato il gene CDKL5 in 273 pazienti con epilessia precoce, con o senza regressione, (213 femmine e 60 maschi) identificando 16 pazienti mutate, con una sensibilità del 7,5%. Fra queste 16, 11 erano portatrici di mutazioni puntiformi, mentre in 5 (1/3 del totale) erano presenti grandi delezioni. I dati relativi alla nostra casistica, così come quelli della letteratura, suggeriscono come la categoria mutazionale delle grandi delezioni sia particolarmente rappresentata, sottolineando l’importanza di completare sempre l’iter diagnostico molecolare sia con il sequenziamento che con l’MLPA. Per il completamento dell’iter diagnostico e per la valutazione della patogenicità della mutazione sia nel caso delle mutazioni di splicing, sia delle delezioni di interi esoni si può rendere necessario analizzare il trascritto del paziente (per questo è necessario un ulteriore prelievo). L’analisi, specie per le mutazioni di splicing, è indispensabile per comprendere la patogenicità del difetto riscontrato e in tutti i casi per definire la mutazione con un nome preciso.
Fra i pazienti con grandi delezioni in letteratura sono stati identificati anche 5 pazienti maschi. Ciò evidenzia come, oltre alla presenza di mutazioni puntiformi, anche la completa assenza (i maschi avendo un solo cromosoma X non presentano una copia non mutata del gene) di regioni estese del gene, non solo sia compatibile con la vita, ma determini un fenotipo paragonabile a quello osservato nelle femmine. In tal senso la minor frequenza osservata di mutazioni nei pazienti maschi (circa il 5% fra i soggetti con encefalopatia epilettica, rispetto al 14% nelle femmine)(Shwann Linag 2011), potrebbe essere dovuta semplicemente al fatto che la sindrome di Rett è classicamente considerata una patologia che colpisce le bambine e quindi vi sia alla base una parzialità o pregiudizio clinico nel suggerire l’analisi di CDKL5 ai maschi con fenotipo Hanefeld, col risultato che un numero minore di maschi vengono identificati.
Sono inoltre stati osservati anche alcuni pazienti a mosaico, cioè in cui solo una quota di cellule presentano l’alterazione genetica in CDKL5, sia con mutazione puntiforme (Masliah-Plachon 2010), sia con grandi delezione (Bartnik 2011), in entrambi i generi. Questo succede perché in tali pazienti la mutazione è il risultato di un evento post-zigotico, cioè non ereditato né insorto nei gameti dei genitori, ma generatosi solo in alcune cellule dell’embrione in stadi precoci del suo sviluppo. Ci si aspetterebbe che tali pazienti abbiano un quadro clinico più sfumato, tuttavia la correlazione è difficile, soprattutto nelle femmine dove alla variabilità tissutale dovuta al grado di mosaicismo, si aggiunge anche quella che dipende dal pattern di inattivazione del cromosoma X, che può variare da sangue periferico al tessuto cerebrale.
Fra i pazienti deleti, circa un terzo, mostrano delezioni estese che interessano anche geni adiacenti a CDKL5. Valutare la estensione precisa della delezione con tecniche aggiuntive, quali per es. l’array-CGH/SNP array, può essere importante (laddove l’MLPA indichi per esempio il coinvolgimento dei primi o ultimi esoni del gene e non dia indicazioni precise sul limite prossimale e/o distale della delezione) perché il coinvolgimento di altri geni può determinare l’insorgenza di manifestazioni cliniche aggiuntive da controllare o monitorare.
Nell’ambulatorio di Genetica Clinica dell’Istituto Auxologico è giunta all’osservazione una paziente di 8 mesi, in cui la delezione identificata tramite analisi MLPA mostrava interessare solo l’esone 1 dell’isoforma I del gene CDKL5, regione che non contiene la sequenza codificante per gli aminoacidi, cioè quella regione che non viene tradotta in proteina, ma che è comunque fondamentale perché contiene le informazioni necessarie all’espressione del gene. L’approfondimento con array-CGH ha evidenziato che la paziente presentava una delezione molto estesa verso il telomero di circa 1,63 Mb, coinvolgente oltre alla regione del promotore di CDKL5 e della regione importante per il legame con MECP2 (Carouge 2010), altri 8 geni oltre a CDKL5. La bambina presentava un quadro compatibile con la variante Hanefeld, con un periodo perinatale non propriamente normale, perché caratterizzato dalla insorgenza di crisi a partire dai tre mesi di vita, ipotonia severa, decelerazione della circonferenza cranica dai 3 mesi, limitato uso delle mani e stereotipie manuali, ridotto contatto oculare e limitata interazione sociale. Inoltre presentava tratti dismorfici con viso triangolare, fronte ampia, orecchie ad impianto basso e anteverse/prominenti e microftalmia. Alcuni di tali tratti dismorfici e la presenza di cataratta in un occhio possono essere riconducibili al fatto che la delezione comprende il gene NHS, responsabile della sindrome di Nance-Horan, sindrome a trasmissione X linked dominante, che si può manifestare anche nelle femmine, seppure in forma più lieve che nei maschi, ed è associata ad anomalie oculari e dentali, facies dismorfica e ritardo mentale nel 30% (dei maschi). Inoltre la bambina presentava un difetto cardiaco, il difetto del setto interatriale, caratterizzato da un’apertura fra il setto che separa i due atri del cuore, reso più particolare e raro dal fatto che l’apertura era doppia. La tetralogia di Fallot similmente ad un altro paziente maschio descritto in letteratura, con delezione paragonabile a quella della nostra paziente (Van Esch, 2007) e recante anch’esso un altro difetto cardiaco potrebbe essere ascrivibile alla delezione di tre geni localizzati nella regione deleta: RAI2, forse target dell’acido retinoico, importante nello sviluppo e patterning del cuore, e SCML1, SCML2, coinvolti nella repressione trascrizionale dei geni HOX, fattori chiave durante lo sviluppo embrionale e si è recentemente visto anche nella formazione del cuore (infatti topi in cui viene eliminato il gene Hoxa1 mostrano anomalie cardiache; Makki, 2012). è attualmente in corso la valutazione dell’espressione quantitativa di CDKL5 in questa bambina per verificare che nonostante la delezione coinvolga solo il prima esone l’espressione sia compromessa.
FORME A VARIANTE CONGENITA e GENE FOXG1
Nel 2005 viene descritta una paziente con grave disabilità intellettiva, agenesia del corpo calloso, difetti di mielinizzazione parietale e frontale nell’insula, microcefalia e dismorfismi, grave ipotonia, ritardo psicomotorio, assenza di linguaggio, forte interazione con lo sguardo, convulsioni cerebrali e tetraplagia. La paziente è portatrice di una traslocazione bilanciata t(2;14)(p22;112) e fiancheggiante un’inversione di 720 kb in 14q12, de novo, che comprende parte di un fattore di trascrizione, FOXG1B (Shoichet et al, 2005). Studi sul modello animale e altri vertebrati dimostrano un’associazione genetica diretta tra il fenotipo del paziente e quello del modello di topo ko (privo di una delle due copie del gene) Foxg1 di topo. Successivamente sono stati identificati mediante array-CGH altri 4 casi con delezione de novo che includevano FOXG1 (Bisgaard et al, 2006). Nel 2008 il gruppo della prof Renieri, in seguito allo studio eseguito mediante la metodica array CGH di una coorte di pazienti con sospetta sindrome di Rett senza mutazioni nei geni MECP2 e CDKL5, identifica, in una paziente con forma congenita nella regione 14q12, una delezione di circa 3Mb che contiene il gene FOXG1. Il sequenziamento del gene FOXG1 in altre 53 pazienti mette in luce la presenza di 2 pazienti con mutazione puntiforme (Ariani et al, 2008; Papa et al 2008). Le caratteristiche cliniche osservate nelle bambine e la funzione del gene suggerivano FOXG1 come un gene responsabile di una quota pazienti. Ad oggi sono stati descritti 28 pazienti con difetto in FOXG1, tra cui 20 femmine e 8 maschi; in 9 casi si tratta di riarrangiamenti cromosomici ed in 19 di mutazioni puntiformi. Si è venuto sempre più definendo un quadro clinico peculiare associato all’assenza in eterozigosi del gene FOXG1, che in parte è sovrapponibile alla sindrome Rett, ma non del tutto, alimentando il dibattito sulla possibile definizione di un sindrome di FOXG1 (Kortum et al, 2011). Le caratteristiche cliniche condivise con la sindrome di Rett sono il ritardo cognitivo grave con assenza di linguaggio, l’ipotonia, le stereotipie manuali/discinesie, le crisi epilettiche generalizzate, i movimenti a scatti, REG, la microcefalia postnatale severa, disturbi del sonno, ed un lieve ritardo di crescita. Sintomi non presenti nella sindrome Rett che si osservano nei pazienti con mutazioni nel gene FOXG1 sono una facies con dimorfismi peculiare, prevalente in presenza di anomalie cromosomiche, lo scarso contatto oculare, l’aprassia, l’ipogenesia del corpo calloso, un pattern semplificato di pachigiria frontale, movimenti corei formi, distonia e spasticità (Kortum et al 2011).
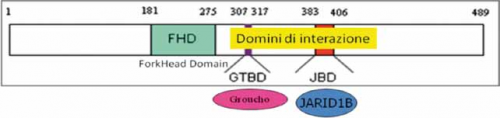
STRUTTURA E FUNZIONE di FOXG1
Il gene FOXG1 codifica per un fattore di trascrizione, denominato Forkhead box protein G1, che viene espresso nel cervello fetale ed adulto e nei testicoli. La proteina interagisce con alcune molecole fondamentali per la regolazione della trascrizione, nello specifico con il repressore trascrizionale JARID1B e con la famiglia delle proteine Groucho che hanno il ruolo di corepressori globali della trascrizione. Questa funzione è di particolare importanza per le fasi precoci dello sviluppo del cervello. Come MeCP2 regola la trascrizione di alcuni geni legandosi indirettamente alle iston-deacetilasi e al DNA metilato (Ariani et al, 2008; Fisher and Caudy, 2008).
Questa proteina è importante durante lo sviluppo embrionale del cervello per la maturazione dei neuroni; si è dimostrato che se presente in quantità ridotta FoxG1 determina una prematura maturazione delle cellule nervose associata ad una riduzione delle dimensioni del telencefalo, (parte più esterna del cervello) e ad un’ipoplasia (limitato sviluppo) del corpo calloso osservato nelle pazienti. Recentemente è stato dimostrato in esperimenti su modelli animali che la proteina FoxG1 non solo è fondamentale per la maturazione embrionale del telencefalo, ma continua ad essere espressa anche nel cervello adulto. Alcuni esperimenti hanno evidenziato come nel cervello adulto di topo, l’espressione indotta di FoxG1 possa bloccare la morte delle cellule nervose ed al contrario la soppressione della sua espressione possa indurne la morte anche in cellule sane. Tali evidenze sostengono l’ipotesi che FoxG1 abbia un ruolo nella sopravvivenza neuronale dei neuroni adulti (Dastidar, 2011). Sempre recentemente è stato studiato il ruolo di FoxG1 a livello di una specifica regione del cervello, il giro dentato ippocampale, dimostrando come: la sua assenza promuova la neurogenesi di neuroni e delle cellule della glia, si produce un numero maggior numero di queste cellule che porta ad un’alterata organizzazione, viene a mancare la corretta stratificazione e come conseguenza si manifesta l’apoptosi (morte) delle cellule stesse.
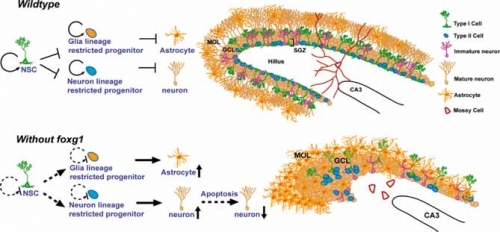 Un ipotetico modello della funzione di Foxg1 nello sviluppo dell’ippocampo
Un ipotetico modello della funzione di Foxg1 nello sviluppo dell’ippocampo
La figura mostra un ipotetico modello della funzione di Foxg1 nello sviluppo dell’ippocampo (area del cervello) postnatale. In presenza di Foxg1, nel giro dentato, una regione del cervello che è rappresentativa dello sviluppo cerebrale, le cellule del sistema nervoso centrale e i precursori continuano a proliferare mantenendo un pool di progenitori normali mentre il corpo cellulare rimane in una regione precisa, strato sub granulare. In mancanza di Foxg1 questo strato viene con una notevole malformazione del giro dentato (Tian et al, 2012).
è importante osservare come l’identificazione di nuove proteine il cui deficit è alla base dello sviluppo di forme atipiche della sindrome di Rett ci permetta di capire meglio le relazioni tra le molecole che guidano la maturazione e lo sviluppo del cervello e che appaiono strettamente correlate. Ciò aggiunge nuovi tasselli per la comprensione dei processi che dal difetto genetico portano alla sintomatologia clinica delle pazienti, ed in futuro al miglioramento dei problemi delle piccole pazienti.
