
La prima giornata del Congresso è stata dedicata ai contributi della genetica alla diagnosi e agli studi sulle correlazioni tra genotipo e fenotipo ad oggi noti.
Sintesi a cura della dott.ssa Silvia Russo
Istituto Auxologico Italiano – Milano
La giornata del 5 giugno 2009 è stata interamente dedicata ai meccanismi genetico-molecolari che sono alla base della Sindrome di Rett e agli studi di modelli murini, importantissimi per comprendere il ruolo e l’interazione reciproca dei geni finora identificati come principali attori della sindrome e per la sperimentazione di approcci terapeutici.
 Momenti del Congresso
Momenti del Congresso
La prima relazione del prof. Michele Zappella, ha fornito un inquadramento storico della sindrome dall’intuizione della sua esistenza, avvenuta negli anni 60, ad opera dell’arguto spirito di osservazione di Andreas Rett e confermata come una nuova sindrome da altri colleghi contemporanei, tra cui il professor Hagerman, che successivamente ha definito il primo consensus di criteri diagnostici per questa malattia. Il riconoscimento definitivo della Sindrome di Rett può essere stabilito nel 1983 con la pubblicazione dei primi 35 casi sugli Annals of Neurology; nel tempo è emersa la grande eterogeneità della presentazione clinica e risale al 1994 da parte di Hagberg e Skjedal la definizione di caratteristiche e criteri che distinguono forme varianti e classiche. Una tappa miliare nella storia della Rett è stata la scoperta, nel 1999, che mutazioni nel gene MeCP2, codificante per la proteina MetilCpG binding 2 fossero responsabili dell’insorgenza della sindrome: questa scoperta ha permesso di dare una diagnosi a tantissime bambine, di spiegare almeno in parte la variabilità fenotipica della malattia, di intraprendere l’ardua strada della comprensione del perché di tutte le specifiche manifestazioni cliniche. Questa proteina oggi sappiamo avere più di un ruolo, è principalmente un regolatore capace di attivare o reprimere l’espressione di molte proteine e per la manifestazione della Sindrome di Rett coinvolge aspetti diversi dello sviluppo fisico e psicomotorio.
La Dott.ssa Pineda ha illustrato le caratteristiche cliniche della sindrome, spiegando come la severità della sua presentazione possa essere valutata utilizzando scale che attribuiscono un punteggio ai vari sintomi clinici, ha descritto nel dettaglio le diverse forme non classiche della malattia e le mutazioni che più frequentemente sono associate ai diversi tipi clinici, forme fruste, encefalopatie congenite, forme con insorgenza precoce delle crisi e forme ad insorgenza tardiva. Studi correlazione tra genotipo e fenotipo attribuiscono la prognosi migliore per l’epilessia alle mutazioni Arg294X, Arg255X e alle delezioni della porzione carbossi-terminale, mentre le pazienti con mutazione Thr158Met sono farmaco resistenti ai farmaci antiepilettici circa 4 volte di più della popolazione generale. Tra le forme atipiche sono state descritte mutazioni missenso, che sostituiscono un singolo aminoacido della proteina MeCP2, quali D151E, Pro127Leu, Pro302Ala, Arg303Ala che non sono incluse tra gli hot-spots mutazionali. Quanto all’incidenza della patologia è pari a circa 1/15000 e si pensa che ogni giorno nel mondo nascano 11 bambine con Sindrome di Rett. Tra le forme non classiche, le pazienti che mostrano un’insorgenza delle convulsioni nel primo anno di vita sono circa il 10% e come ha illustrato la Dott.ssa Haley Archer, dell’Istituto di Genetica Medica di Cardiff, il primo gene per cui sono state descritte mutazioni in pazienti atipiche è il gene CDKL5 (ciclin-dependent kinase-like 5) che codifica per una serin-treonin chinasi e le cui mutazioni sono responsabili proprio di questo tipo di quadro clinico. Secondo i dati riportati dalla Harcher le mutazioni di CDKL5 sono causa dell’8% delle pazienti con encefalopatia precoce, del 17% delle pazienti con spasmi infantili e del 13% delle forme Rett-like. Si tratta di mutazioni che si localizzano lungo tutto il gene, con una maggior presenza di mutazioni puntiformi nel dominio catalitico; le mutazioni sono per lo più private, ossia non ve ne sono di ricorrenti. Sono state riportate recentemente anche ampie delezioni nella porzione amino-terminale del gene. La proteina è espressa, sia nel corpo sia nei dendriti nei neuroni e fosforila MeCp2, è stata dimostrata la loro compresenza di CDKL5 e di MECP2 nei neuroni anche se non in tutti i tessuti del cervello; l’espressione di CDKL5 aumenta dopo la nascita e sembra esprimersi prima in quei neuroni che hanno raggiunto la piastra corticale. La Archer ha descritto anche gli aspetti clinici: le prime crisi insorgono generalmente prima dei 3 mesi, distinguendo 3 periodi, uno iniziale con un normale EEG interictale, un secondo stadio con spasmi infantili (60-70% delle pazienti), encefalopatia precoce e normali EEG interictali ed infine un terzo stadio con epilessia intrattabile e mioclonia. Lo sviluppo psicomotorio appare ritardato da subito con presenza di ipotonia, difficoltà ad alimentarsi, pianto continuo e difficoltà a fissare. Si può dire le bambine con mutazioni in CDKL5 si distinguono dalla forma classica, oltre che per la precoce insorgenza delle crisi per la mancanza di un periodo di vita normale e quindi di regressione, per la mai acquisita capacità di interazione sociale, per l’assenza di interazione sociale, mentre appaiono meno presenti o comunque meno gravi, problematiche come la scoliosi, l’iperventilazione e i disturbi del sistema autonomico. Ulteriori aggiornamenti circa la proteina CDKL5 nel pomeriggio da parte della Dott.ssa Kalsheuer, i cui esperimenti portano alla conclusione che CDKL5, localizzata all’esterno della membrana, si concentri soprattutto nel punti di contatto fra le cellule e appartenga ad un network di proteine che ha il ruolo di creare e regolare i contatti tra le cellule. Dimostrano inoltre l’interazione di CDKL5 con molte proteine che appartengono a classi molecolari diverse, quali i costituenti del citoscheletro, molecole coinvolte nel traffico di vescicole e nel turnover di proteine.
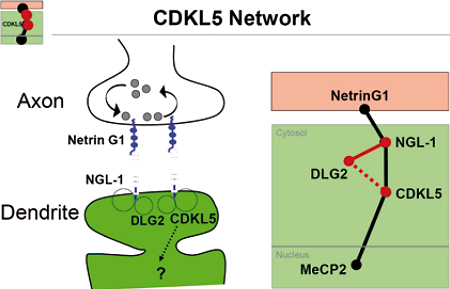
Rapporti tra i geni coinvolti nella Sindrome di Rett e ed interattori di CDKL5
Una ricerca sistematica ha evidenziato 18 possibili interattori di CDKL5, tra cui particolarmente interessante NGL1, una molecola di adesione cellulare neuronale che interagisce con la proteina Nectrina 1, per cui era stata descritta la sua interruzione in seguito ad un riarrangiamento in una paziente con un quadro clinico Rett. Sempre la Kalsheuer riferisce dell’identificazione del nuovo esone 16a di CDKL5 prevalentemente espresso nelle isoforme cerebrali, in cui nei pazienti testati per ora non è stata riportata nessuna mutazione.
La Dott.ssa Mari, Università di Siena, ha riportato i risultati degli screening di mutazioni del terzo gene coinvolto nella Sindrome di Rett, il gene FOXG1 che mappa nella regione cromosomica 14q12. In seguito all’identificazione mediante applicazione della metodica array-CGH, di una delezione di 3Mb nella regione 14q12 in una paziente con caratteristiche Rett-like e lievi tratti dismorfici, è stata analizzato il contenuto genico della regione suggerendo FOXG1 come un candidato plausibile. FOXG1 infatti codifica per un fattore di trascrizione, la proteina “forkhead-box G1 protein”, che si esprime nei neuroni corticali in differenziazione e maturi del cervello fetale e adulto e nei testicoli. Un primo studio mutazionale eseguito a Siena ha identificato due mutazioni in troncanti in una coorte di 53 bambine negative ad un precedente screening di MeCP2/CDKL5 e uno studio europeo (vedi figura) eseguito su 107 pazienti europei tra cui 6 maschi, ha portato all’identificazione di altri 4 casi con mutazione cui si è aggiunta molto recentemente una paziente francese, tutti riconducibili ad un fenotipo di variante congenita.
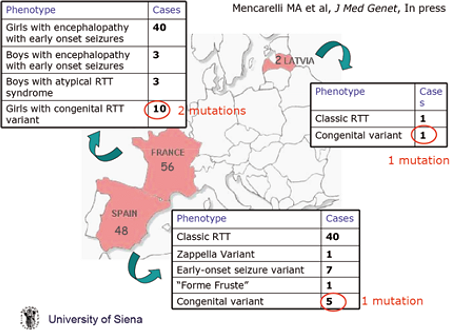
Studio europeo sulle pazienti con mutazioni nel gene FOXG1
Infatti la raccolta di questi casi ha consentito di cominciare a definire il fenotipo clinico delle pazienti con mutazioni in FOXG1: periodo perinatale nella norma caratterizzato da una grave ipotonia, microcefalia prima dei 4 mesi, regressione entro i 6 mesi, assenza dell’uso delle mani e del linguaggio e stereotipie delle mani tipiche della Rett, grave ritardo cognitivo, ipoplasia del corpo calloso, movimenti costanti della lingua, sono presenti movimenti a scatti degli arti superiori, mentre l’epilessia non è molto presente. È stato fatto presente come un fenotipo molto simile, è stato riportato in letteratura in altre pazienti con delezioni della banda 14q12, come se esistesse un fenotipo associato alle delezioni di questa regione e sia quindi consigliabile testare con array-CGH i pazienti negativi allo screening di MeCP2/CDKL5 ed encefalopatia congenita.
L’argomento delle mutazioni di MeCP2 nei soggetti di sesso maschile è stato affrontato dalla Dott.ssa Van Esche, del Centro di Genetica Medica di Leuven in Belgio, che ha descritto la distinzione in tre diverse classi fenotipiche: a) maschi con mutazioni presenti nelle bambine con Rett classica mostrano un’encefalopatia congenita, hanno un ritardo mentale gravissimo e generalmente non sopravvivono oltre la prima infanzia, b) maschi con un quadro clinico Rett-like, generalmente con un genotipo Klinefelter o a mosaico, c) un gruppo eterogeneo di maschi con ritardo mentale da moderato a severo che presenta mutazioni, mai descritte nelle bambine come patogenetico e generalmente ereditate dalle madri sane. Un’ulteriore classe di maschi con difetti genetici in MeCP2 è rappresentato da duplicazioni che coinvolgono il gene MeCP2, per un’incidenza pari all’1% se si considera un gruppo di soggetti maschi con ritardo mentale, ma sale fino al 2% in maschi con encefalopatia congenita. Le duplicazioni hanno un’estensione che varia da 100 a 900 Kb e includono almeno i geni MeCP2 e IRAK. Le caratteristiche cliniche comuni sono ipotonia infantile, spasticità progressiva, convulsioni, assenza di linguaggio e infezioni ricorrenti.
Nelle comunicazioni orali del pomeriggio sono stati presentati studi volti a meglio comprendere dove operano i geni coinvolti nella Sindrome di Rett, eventuali target e modificatori coinvolti nel modulare l’attività dei geni stessi. La Dott.ssa Sara Ricceri, Divisione di Neuroscienze dell’Ospedale San Raffaele, ha riportato studi sulla localizzazione di CDKL5 negli spicchi nucleari. Gli spicchi nucleari sono un sito di conservazione di fattori di splicing, chinasi, fosfatasi e proteine strutturali e sono regolati da meccanismi di fosforilazione e defosforilazione. Recenti esperimenti sembrano dimostrare che un’espressione maggiore del normale della proteina CDKL5 disaggrega gli spicchi nucleari, e che mutazioni ne influenzino la struttura. Si suggerisce che CDKL5 sia associato agli spicchi nucleari e sia necessario per la loro coalescenza, che regoli la loro struttura e che sia coinvolto nella attività di formazione dell’mRNA e splicing alternativo. Ciò vuol dire che CDKL5 è importante per il corretto assembramento del codice che porterà alla formazione di proteine corrette. La Dott.ssa Kilstrup-Nielsen ha mostrato dati interessanti sulla proteina HIPK2 (Homeodomain interacting protein kinase 2). Questa proteina è una serin treonin chinasi originariamente identificata come repressore di fattori di trascrizione degli omodomini, è espressa ubiquitariamente, ma principalmente nel sistema nervoso. Il topo KO presenta ritardo psicomotorio e si è dimostrata una perdita selettiva di neuroni dopaminergici. Perché HIPK2 è coinvolta nella Sindrome di Rett? Esperimenti di immunoprecipitazione hanno dimostrato che HIPK2 si associa in vivo a MeCP2 ed che è in grado di determinare la sua fosforilazione endogena in vitro a livello dello specifico aa Ser80 e che insieme contribuiscano a indurre l’apoptosi cellulare ossia la morte delle cellule. Possiamo considerare HIPK2 un modulatore dell’attività di MeCP2.
 I partecipanti durante una pausa nel Chiostro dell’Università Statale di Milano, sede del Congresso
I partecipanti durante una pausa nel Chiostro dell’Università Statale di Milano, sede del Congresso
L’ultima parte della giornata è stata dedicata allo studio di modelli murini e gli approcci terapeutici che si stanno sperimentando. A questo proposito il Dott. Roux dell’università di Marsiglia, ha spiegato come i difetti di respirazione siano presenti nel topo dal primo mese di vita e aumentino intorno al secondo mese fino ad essere causa di morte. Questo ha permesso di studiarne le cause. Si osserva una diminuzione dei neuroni catecolaminergici in siti specifici del sistema nervoso centrale, medulla e locus ceruleo. Si pensa che la tirosinidrossilasi o un’altra molecola del pathway siano target d MeCP2. Cercando un trattamento farmacologico che alleviasse i problemi respiratori, si è osservato che la desapramina stabilizza la respirazione e allunga i tempi di vita. Dopo avere sperimentato sul modello murino il trattamento, ora è in corso in Francia una sperimentazione su 36 bambine Rett di età compresa tra i 4 e 18 anni. Il goal sarà quello di identificare nuove molecole da provare sulle bambine per migliorare il maggior numero possibile di sintomi.
Successivamente il Dott. Eubanks, università di Toronto, ha illustrato i suoi studi sui recettori NMDA (N-metil-d- aspartato) critici per la maturazione delle sinapsi e la formazione della plasticità sinaptica. Partendo dal presupposto che la maturazione delle sinapsi è alterata nella Sindrome di Rett hanno voluto osservare il livello di espressione dei recettori NMDA o la loro localizzazione fosse alterato nel cervello dei topi meCP2y/- dimostrando che effettivamente si ha una dimunizione del numero di recettori, ma la localizzazione non viene alterata dimostrando che la sinaptogenesi è alterata ma in modo sottile. Sempre Eubanks ha fornito successivamente una carrellata descrittiva dei principali modelli murini e della loro importanza nella comprensione dei meccanismi di base della sindrome e per lo sviluppo di approcci terapeutici. Ha ricordato il modello di Adrian Bird in grado di revertire la malattia, sarebbe importante avere modelli murini specificamente corrispondenti alle diverse mutazioni, perché come l’espressione della malattia varia secondo il genotipo, così ci si può aspettare anche la risposta ad un eventuale trattamento terapeutico.
Ha soffermato quindi la sua attenzione su quelle molecole in grado di correggere le mutazioni di stop, superando l’errore e continuando la traduzione della proteina; il successo di questo meccanismo è condizionato anche dal tipo di sequenza che circonda la mutazione. Tra queste molecole è stata sperimentata la gentamicina e la molecola PTC124. Restano importanti problemi da risolvere:
- la tossicità del farmaco, per cui potrebbero essere necessarie dosi troppo elevate per riuscire ad ottenere un effetto soddisfacente;
- la funzionalità della proteina corretta potrebbe essere limitata o troppo ridotta la loro quantità. A questo scopo è necessario sviluppare nuovi modelli murini e diversi laboratori nel mondo sono impegnati a questo scopo.
