Al Convegno di Trento, sono stati presentati in anteprima i risultati di uno studio sul modello animale per la Sindrome
di Rett, condotto da membri dell’AIRett Research Team (Giovanni Laviola e Bianca De Filippis, del Dipartimento di Biologia cellulare e Neuroscienze dell’Istituto Superiore di Sanità). Tale studio (De Filippis et al., 2014) dimostra il potenziale terapeutico di un trattamento farmacologico innovativo, basato sulla stimolazione di un recettore per la serotonina nel sistema nervoso centrale. Grazie all’integrazione di competenze multidisciplinari (neuroscienze comportamentali e neurobiologia), e per verificare la loro ipotesi, lo studio si è avvalso di LP-211, una molecola agonista selettivo del recettore 7 per la serotonina, recentemente sintetizzata dal gruppo di chimica-farmaceutica del Prof. M. Leopoldo dell’Università di Bari. I ricercatori sono riusciti a dimostrare come la somministrazione di LP-211 è in grado di ripristinare alcuni processi molecolari responsabili della plasticità e morfologia delle cellule nervose, deficitari nel cervello dei topi mutanti per Mecp2. Allo stesso tempo, sono stati contrastati efficacemente alcuni dei sintomi caratteristici della sindrome, come l’elevata emotività, i problemi cognitivi e i deficit di coordinazione motoria.
Studi recenti suggeriscono un ruolo chiave per l’insorgenza dei suddetti sintomi nell’incapacità delle cellule del sistema nervoso di scambiarsi correttamente le informazioni. Ad attrarre in modo particolare l’attenzione dei ricercatori è stato il ruolo importante della serotonina nella regolazione di un ampio spettro di meccanismi neurofisiologici che appaiono alterati in sindromi dello spettro autistico e nella Rett.
La serotonina (5HT), come altri neurotrasmettitori, veicola l’informazione tra specifici neuroni e svolge la sua azione legandosi a diversi recettori. La serotonina è coinvolta in un’ampia varietà di processi che avvengono nel sistema nervoso centrale, inclusi la regolazione del comportamento alimentare, il ritmo sonno-veglia, la socialità, il tono dell’umore, l’emotività e i processi cognitivi. È noto che esistono sette classi principali di recettori che mediano gli effetti della serotonina nel sistema nervoso. Il recettore 7 (5-HT7) della serotonina, quello identificato più di recente, è presente nel cervello con elevata densità nel talamo e nell’ipotalamo, così come nell’ippocampo e nella corteccia. È stato recentemente possibile stabilire una chiara relazione tra il recettore 5-HT7 e fenomeni neurofisiologici quali regolazione dei ritmi circadiani, sonno, il tono dell’umore e la termoregolazione. Il ruolo dei recettori 5-HT7 è attualmente studiato nell’ambito di diverse patologie neurologiche e psichiatriche. Inoltre, un numero crescente di studi ha evidenziato un ruolo chiave anche nei processi cognitivi, nella neurogenesi, nella
regolazione fine della morfologia neureonale e infine nella funzione e plasticità sinaptiche.
Studi recenti hanno dimostrato che la neurotrasmissione monoaminergica e della serotonina è deficitaria sia nei pazienti RTT che in modelli murini della malattia, suggerendo un nuovo bersaglio terapeutico per curare la Sindrome di Rett. I risultati presentati dimostrano che l’attivazione di un particolare sottotipo recettoriale della serotonina, il recettore 5-HT7, mediante l’utilizzo di un agonista selettivo, ripristina i difetti neuronali e comportamentali associati alla mutazione di MeCP2.
La stimolazione di questo specifico recettore attiva una serie di processi intracellulari, tra cui anche le GTPasi della famiglia Rho. L’attivazione farmacologica di queste vie molecolari nella cellula ha come conseguenza di promuovere la plasticità nervosa.
Le Rho GTPasi sono proteine espresse in modo ubiquitario nelle cellule eucariotiche che agiscono da interruttori in diversi tipi di trasmissione del segnale. Fra le numerose funzioni svolte dalle RhoGTPasi, di particolare interesse è il ruolo fondamentale che esse svolgono nel mediare i cambiamenti strutturali e morfologici neuronali, la connettività e la funzionalità della glia in seguito a stimoli esterni.
Dato il ruolo chiave svolto da questa famiglia di proteine nel controllare la plasticità neuronale, è plausibile che anomalie nel loro funzionamento possano essere almeno in parte responsabili della ridotta plasticità sinaptica che caratterizza la RTT. Ci siamo quindi chiesti se un intervento farmacologico che agisse anche a livello delle RhoGTPasi, potesse rivelarsi la giusta strategia per contrastare, almeno in parte, la sintomatologia RTT. A sostegno di questa ipotesi è il fatto che molte forme di disabilità intellettiva non sindromica sono caratterizzate da anomalie morfologiche neuronali, in particolare nei dendriti e nelle spine, che sono note essere associate ad alterazioni del funzionamento
delle RhoGTPasi. In particolare, in linea con le evidenze della letteratura, uno studio di De Filippis e colleghi, pubblicato nel 2012, dimostrava che la somministrazione intracerebrale della tossina batterica CNF1, attivava specificamente le RhoGTPasi cerebrali e contrastava efficacemente i deficit di apprendimento e la ridotta plasticità sinaptica presente in topi mutanti per Mecp2.
Per verificare la potenziale efficacia del trattamento con LP-211 nel contrastare alcuni dei sintomi caratteristici della RTT ci si è quindi avvalsi di un modello di topo transgenico per la RTT (Mecp2308) in una fase pienamente sinto-
matica, sfruttando la conoscenza approfondita delle alterazioni neurobiologiche e comportamentali acquisita in precedenza, che si ritiene siano riconducibili ai sintomi mostrati dalle pazienti.
3a. Il comportamento
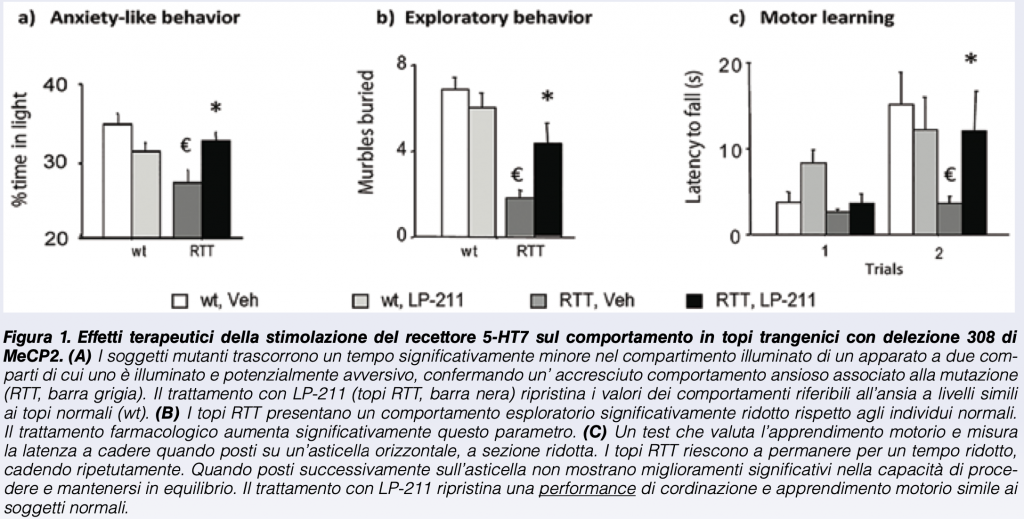
I risultati dello studio presentato hanno evidenziato, a seguito della somministrazione ripetuta con LP-211, un significativo miglioramento in diversi domini comportamentali. Il trattamento con LP211 ha infatti significativamente alleviato gli elevati livelli di comportamento ansioso e migliorato i deficit di coordinazione motoria e di esploratività dei topi mutanti (Figura 1).
Oltre ad esercitare effetti benefici sulle capacità motorie, LP-211 ha determinato una significativa influenza nel contrastare i deficit di abilità cognitive presenti nel modello per la RTT.
3b. La neurobiologia
La valutazione del ruolo del citoscheletro (struttura e morfologia della cellula) ha avuto recentemente un ampio sviluppo nello studio della eziopatogenesi della Sindrome di Rett. In questo ambito, l’analisi a livello cerebrale consentita, è importante in questa sede ricordarlo, dalla disponibilità
me di disabilità intellettiva: PAK e rpS6, molecole coinvolte nella regolazione e nella sintesi delle pro-
fluenza nel contrastare i deficit di abilità cognitive presenti nel modello per la RTT.
3b. La neurobiologia
La valutazione del ruolo del citoscheletro (struttura e morfologia della cellula) ha avuto recentemente un ampio sviluppo nello studio della eziopatogenesi della Sindrome di Rett. In questo am-
di un modello animale per RTT, ha evidenziato una diminuzione nella densità del recettore 5-HT7 a livello della corteccia e dell’ippocampo nei topi RTT. Alterazioni importanti sono state inoltre evidenziate nei meccanismi molecolari responsabili per la morfologia e struttura della cellula e della plasticità sinaptica e già note per il loro coinvolgimento in diverse forme di disabilità intellettiva: PAK e rpS6, molecole coinvolte nella regolazione e nella sintesi delle proteine strutturali della cellula. Tali alterazioni di base hanno subito un drammatico miglioramento in seguito al trattamento con LP-211.
Studi precedenti condotti in vitro hanno dimostrato il coinvolgimento attivo di questo recettore nella regolazione della morfologia della cellula nervosa e nella plasticità sinaptica. Tale azione si ritiene mediata dalla stimolazione selettiva di alcuni attivatori molecolari della famiglia delle Rho GTPases, RhoA and Cdc42, che rivestono un ruolo importante nella regolazione della struttura della cellula (citoscheletro) nervosa. Alterazioni a livello delle Rho GTPases sono state evidenziate in diverse forme di disabilità intellettiva e danno luogo a deficit sia di connettività tra le aree cerebrali sia di capacità cognitive. Nonostante il ruolo critico svolto dalle Rho GTPases nella corretta funzionalità cellulare, al momento sono veramente poche le molecole farmacologiche disponibili per una loro modulazione terapeutica.
Questi risultati suggeriscono che alcuni dei sintomi caratteristici della Sindrome di Rett possano essere efficacemente contrastati dalla stimolazione selettiva del recettore 7 della serotonina e la conseguente attivazione farmacologica delle RhoGTPasi. Queste proteine responsabili della plasticità cellulare vengono identificate quindi come un promettente target innovativo per la terapia e la diagnostica della patologia. LP211 sembrerebbe inoltre rappresentare una molecola ad azione promettente, da sviluppare come farmaco, per il contrasto della grave sintomatologia che colpisce le pazienti in una fase avanzata della sindrome.
Nel quadro di questo studio, si ritiene che le scoperte relative alla reversibilità del fenotipo RTT aprano la prospettiva di una terapia di tipo non genico, suggerendo anche la possibilità che la plasticità neuronale e i deficit cognitivi possano essere migliorati attraverso dei trattamenti farmacologici che stimolino la connettività neuronale.
Data l’estrema complessità del quadro clinico e l’assenza di terapie mirate, anche effetti solamente parziali del trattamento con LP-211 sulla sintomatologia RTT sembrano di particolare rilevanza. Inoltre, poiché la stimolazione del recettore 5-HT7 induce l’attivazione a livello cellulare della famiglia delle RhoGTPasi, ampiamente coinvolte nei fenomeni di plasticità, e il cui coinvolgimento nelle sindromi di disabilità intellettiva su base genetica è ben noto, l’evidenza di possibili effetti terapeutici potrebbe essere estesa ad altre sindromi di disordini del neurosviluppo.
I risultati ottenuti in questo studio, pubblicato nella prestigiosa rivista scientifica Neuropsychopharmacology (De Filippis et al. 2014), sono estremamente incoraggianti in quanto aprono la strada alla possibilità di un’applicazione terapeutica di questa nuova molecola nella Sindrome di Rett, ma anche in altre patologie altamente invalidanti della popolazione pediatrica, caratterizzate da deficit cognitivi e motori. Di nota ancora molta strada deve essere percorsa in questa direzione prima che questo potenziale approccio terapeutico possa essere valutato per la clinica.
Al fine di identificare una soluzione per cercare di ridurre i difetti della traduzione proteica, abbiamo cercato la causa del danno alla base del suo malfunzionamento. Il risultato di questa ricerca è stato quello di individuare una via di processi del funzionamento della cellula quale causa diretta dei difetti neurologici. Sono infatti coinvolti la sintesi di nuove proteine, che regola le dimensioni e la forma dei neuroni, cosi come la morfologia ed il funzionamento delle sinapsi, i parametri neurologici che sono alterati nella Sindrome di Rett. Il malfunzionamento nelle cellule nervose è noto in molte altre gravi patologie neurologiche dello sviluppo accompagnate da disabilità intellettiva grave come l’X-Fragile. Questi studi indicano che sono stati identificati meccanismi molecolare chiave per il corretto funzionamento dei circuiti nervosi e che cominciamo a modularli efficacemente, aprendo in qualche modo la via per scoprire delle cure efficaci contro il malfunzionamento presente non solo nelle bambine Rett ma anche in altre patologie del neurosviluppo.
Lo studio è stato finanziato parzialmente da AIRETT, Italia e dalla Fondation Jerome Lejeune, Francia. I ricercatori hanno già registrato un primo brevetto e sperano ora di trovare investitori che desiderino puntare sullo sviluppo ulteriore del farmaco, per poter arrivare nel minor tempo possibile alla sperimentazione sull’uomo.