
Aglaia Vignoli, Centro Epilessia, AO San Paolo, Dipartimento Scienze della Salute, Università di Milano
L’epilessia è una delle problematiche di maggiore impatto clinico nelle pazienti con Sindrome di Rett. Generalmente viene riportato che nella storia naturale della sindrome, nelle pazienti adulte le crisi epilettiche tendono a diminuire e/o a scomparire.
Recenti studi su ampie casistiche di pazienti con Sindrome di Rett confermano queste ipotesi: uno studio condotto da medici australiani su 685 pazienti dimostra che nelle pazienti di età superiore ai 17 anni solo il 40% ha ancora crisi, il 23% a frequenza settimanale. Per quanto riguarda una possibile correlazione genotipo-fenotipo le mutazioni con maggior rischio di farmacoresistenza sono risultate: p. T158M e Large deletions. I farmaci più utilizzati in questa ampia casistica erano: acido valproico (47%), carbamazepina (39%), lamotrigina (30%), levetiracetam (24%), topiramato (19%) (Bao et al., 2013).
Un altro studio sull’epilessia condotto in Israele su 97 pazienti, ha individuato come criterio prognostico favorevole sul controllo delle crisi, un’età di esordio più tardiva (dopo i 5 anni) (Nisserkon et al., 2010).
I dati dei colleghi americani sulla storia naturale dell’epilessia nella Sindrome di Rett in 360 pazienti confermano che dopo la pubertà la ricorrenza delle crisi tende a diminuire e il 36% delle pazienti è libera da crisi. La mutazione associata a maggior rischio di epilessia è T158M, a minor rischio R255X e R306C (Glaze et al., 2010).
Anche nella casistica italiana, l’epilessia dopo l’adolescenza tende a essere meglio controllata: il 41% delle pazienti infatti risulta libera da crisi, tra queste nel 7% dei casi è stato possibile sospendere la terapia; in coloro che ancora hanno crisi il 16% ha crisi sporadiche, mentre il 36% crisi farmacoresistenti (Vignoli et al., 2011).
I dati provenienti dalla popolazione Rett dell’Olanda (37 pazienti seguite nel tempo) dimostrano che in età adulta il 54% delle pazienti è libera da crisi, con una stabilizzazione e/o miglioramento dell’epilessia fra 20-30 anni. Inoltre viene segnalato un miglioramento delle funzioni cognitive con l’età (Halbach et al., 2013).
Abbiamo voluto caratterizzare meglio dal punto di vista epilettologico la casistica seguita presso l’Azienda Ospedaliera San Paolo di Milano; dal 2005 ad oggi, sono state seguite 92 pazienti con Sindrome di Rett, di cui 60 bambine (12 mesi-17 anni) e 32 adulte (19-42 anni).
Delle pazienti adulte, 25 presentano una Sindrome di Rett classica, 4 la variante a linguaggio preservato, 2 la variante con convulsioni precoci, 1 la forma congenita. Tutte hanno eseguito indagine genetica per MECP2 ed in 28 è stata trovata la mutazione (87%).
Per quanto riguarda le caratteristiche strettamente epilettologiche, 28/32 hanno una diagnosi di epilessia, mentre una paziente ha presentato un unico episodio critico in febbre.
L’età di esordio più frequentemente riportata era fra i 2 e i 9 anni, 4 pazienti hanno esordito nei primi 18 mesi di vita, 5 in età adolescenziale, una sola in età adulta (20 anni) (Figura 1).
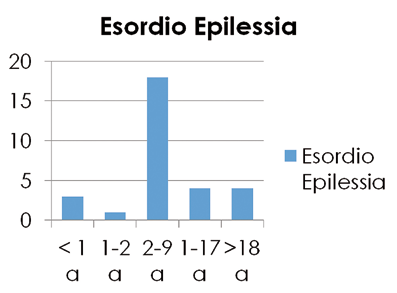 Figura 1: età di esordio delle crisi
Figura 1: età di esordio delle crisi
Le caratteristiche delle crisi all’esordio erano così distribuite: 13 crisi focali, 8 crisi generalizzate tonico-cloniche, 3 assenze atipiche, 2 crisi toniche, 1 crisi miocloniche ed in 1 spasmi (Figura 2).
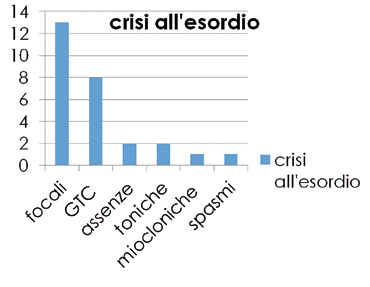
Mentre al follow-up, solo il 50% delle pazienti aveva ancora crisi attive, in 4 pazienti a frequenza sporadica, mentre 10 pazienti hanno una epilessia farmacoresistente che richiede una politerapia (acido valprico, lamotrigina, carbamazepina, levetiracetam).
Le caratteristiche delle crisi al follow-up mantengono una prevalenza di crisi focali come all’esordio (Figura 3).
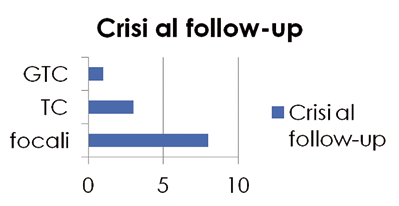
La restante metà del campione (14 pazienti) era libera da crisi (10 pazienti da più di 5 anni), in 3 casi le pazienti hanno sospeso terapia; in 11 proseguono una monoterapia (acido valproico, lamotrigina, carbamazepina).
Esaminando invece le caratteristiche neurologiche e comportamentali del campione, abbiamo rilevato che 15 camminano autonomamente, 10 con sostegno, le restanti hanno perso questa abilità. Per quanto riguarda le prassie delle mani: 6 pazienti hanno conservato un parziale utilizzo delle mani (toccano, afferrano, portano alla bocca); tutte le pazienti presentano delle stereotipie anche se meno intense rispetto all’infanzia.
Rispetto alle manifestazioni osteoarticolari: tutte presentano scoliosi e rigidità articolare.
Frequenti sono i disturbi del comportamento (in genere crisi di agitazione psicomotoria): 10 pazienti assumono per questa indicazione una terapia farmacologia (Risperidone, acido valproico, Trittico); inoltre 11 pazienti hanno un disturbo del sonno (insonnia, risvegli frequenti).
In conclusione, sia i dati della letteratura internazionale, sia la nostra esperienza personale confermano che l’epilessia nella Sindrome di Rett generalmente tende a stabilizzarsi in età adulta. Per tale motivo può essere utile in accordo con la famiglia, dopo valutazione attenta, riconsiderare la terapia antiepilettica assunta per eventuali modifiche e/o sospensione. Al di là del dato epilettologico, i disturbi del sonno e problemi comportamentali si mantengono in età adulta.
D’altra parte migliorano le funzioni cognitive (attenzione, intenzionalità/competenze relazionali e comunicative), forse anche in relazione al miglior controllo delle crisi.
Bibliografia:
Bao X, Downs J, Wong K, Williams S, Leonard H. Using a large international sample to investigate epilepsy in Rett syndrome. Dev Med Child Neurol. 2013 Jun;55(6):553-8. doi: 10.1111/dmcn.12093. Epub 2013 Feb 19. PubMed PMID:23421866.
Glaze DG(1), Percy AK, Skinner S, Motil KJ, Neul JL, Barrish JO, Lane JB, Geerts SP, Annese F, Graham J, McNair L, Lee HS. Epilepsy and the natural history of Rett syndrome. Neurology. 2010 Mar 16;74(11):909-12. doi: 10.1212/WNL.0b013e3181d6b852.
Halbach NS, Smeets EE, Steinbusch C, Maaskant MA, van Waardenburg D, Curfs LM. Aging in Rett syndrome: a longitudinal study. Clin Genet. 2013 Sep;84(3):223-9.doi: 10.1111/cge.12063. Epub 2012 Dec 7. PubMed PMID: 23167724.
Nissenkorn A, Gak E, Vecsler M, Reznik H, Menascu S, Ben Zeev B.Epilepsy in Rett syndrome—the experience of a National Rett Center. Epilepsia. 2010 Jul;51(7):1252-8. doi: 10.1111/j.1528-1167.2010.02597.x. Epub 2010 May 13.
Vignoli A, La Briola F, Peron A, Turner K, Savini M, Cogliati F, Russo S, Canevini MP. Medical care of adolescents and women with Rett syndrome: an Italian study. Am J Med Genet A. 2012 Jan;158A(1):13-8. doi: 10.1002/ajmg.a.34367. Epub 2011 Dec
