
Tommaso Pizzorusso
Istituto di Neuroscienze del CNR, Pisa e Dipartimento di Psicologia, Università di Firenze
Sebbene le mutazioni genetiche responsabili della sindrome di Rett siano sempre più note, le alterazioni a livello cellulare che queste mutazioni producono sofigurano ancora oggetto di intenso studio. La conoscenza di queste alterazioni cellulari e delle vie molecolari che le determinano è di fondamentale importanza in quanto è responsabile della sintomatologia che si osserva nel paziente e nei modelli animali. Inoltre, queste vie molecolari implicate sono potenzialmente bersaglio di trattamenti farmacologici capaci migliorare i sintomi.
Studi recenti suggeriscono che cellule non neuronali, quali gli astrociti o la microglia1,2, possano giocare un ruolo molto importante nello scatenarsi della sintomatologia nel modello di mutante per MeCP2. Tuttavia è logico supporre che il manifestarsi dei sintomi sia a carico di alterazioni neuronali indotte primariamente nel neurone stesso o conseguenza di disfunzione a carico dell’astroglia o della microglia. Di conseguenza, risulta fondamentale lo studio delle connessioni neuronali in presenza di mutazioni responsabili di Sindrome di Rett.
Gli studi finora condotti nei modelli di topo e nei pazienti hanno mostrato alterazioni principalmente a carico delle sinapsi, consistenti in una riduzione della densità e della funzione delle sinapsi eccitatorie e inibitorie3. Le spine dendritiche costituiscono una zona specializzata del neurone su cui si localizzano molti ingressi sinaptici; principalmente di tipo eccitatorio, sebbene recenti studi suggeriscano che anche le sinapsi inibitorie possano formarsi a livello delle spine. Numerosi studi hanno dimostrato una riduzione della densità delle spine dendritiche in diverse aree cerebrali e in diversi modelli murini di Sindrome di Rett3, suggerendo quindi che le alterazioni delle spine siano un aspetto ricorrente della patologia. In effetti, anche gli studi nei pazienti hanno mostrato alterazioni a livello delle spine dendritiche4. Per quanto importanti, tuttavia, questi studi non danno informazioni sui processi cellulari la cui alterazione determina la riduzione delle spine dendritiche5. Ciascuna spina dendritica, infatti, va incontro a un processo di maturazione che prevede un’iniziale formazione di una spina immatura (il cosiddetto filopodio) che può essere ritratto oppure selezionato per il successivo stadio di maturazione che porta alla formazione della spina matura. A sua volta la spina matura può essere ritratta oppure andare incontro a una stabilizzazione che le permetterà di permanere per mesi o per anni nel circuito neuronale.
È evidente che ciascuno di questi processi avrà alla base diversi meccanismi molecolari e che una riduzione della densità delle spine potrebbe derivare da disfunzioni di ciascuno di questi processi. Per esempio, si potrebbe ipotizzare sia una mancata formazione che un’accelerata retrazione come meccanismo di base di una riduzione della densità delle spine dendritiche.
Per risolvere questo problema è necessario, quindi, utilizzare tecniche che permettano di seguire la vita delle spine dendritiche dalla loro formazione alla loro retrazione o stabilizzazione finale. Queste metodiche sono disponibili da circa dieci anni e solo negli ultimi due anni sono state applicate all’analisi dei modelli animali della Sindrome di Rett6. La microscopia a 2 fotoni permette, infatti, di osservare ripetutamente per ore fino a mesi, in vivo, le spine dendritiche rese fluorescenti per mezzo di opportune tecniche genetiche.
Presso il National Enterprise for Science and Technology (NEST) della scuola normale superiore di Pisa nel laboratorio coordinato dal Dr. Gimmi Ratto, in collaborazione con l’istituto di neuroscienze del CNR di Pisa (Prof. Tommaso Pizzorusso e Dr.ssa E.M. Boggio) e del Dipartimento di anatomia, farmacologia e medicina legale dell’Università di Torino (Prof. Maurizio Giustetto), il gruppo di ricerca guidato dalle Dr.sse Silvia Landi e Elena Putignano ha analizzato la formazione e la maturazione delle spine dendritiche in topi con delezione di MeCP26. Le analisi sono state effettuate in topi di quattro settimane, ovvero un’età che precede il manifestarsi della maggior parte dei sintomi. Mediante l’osservazione al microscopio due fotoni delle spine dendritiche dei neuroni della corteccia cerebrale, abbiamo verificato una drammatica riduzione del tasso di produzione di nuovi filopodi nei neuroni portatori della delezione di MeCP2.
Inoltre, i pochi filopodi che si formavano in assenza di MeCP2, non mostravano quei caratteristici movimenti che portano alla successiva maturazione della sinapsi (Fig.1).
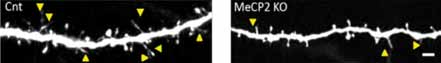 Figura 1. Esempio di un dendrite di controllo (CrT) o di topo con delezione di MeCP2 (MeCP2 KO). Le frecce gialle indicano i filopodi. Notare la scarsità di filopodi nel topo mutante per MeCP2
Figura 1. Esempio di un dendrite di controllo (CrT) o di topo con delezione di MeCP2 (MeCP2 KO). Le frecce gialle indicano i filopodi. Notare la scarsità di filopodi nel topo mutante per MeCP2
Questo primo risultato porta ad individuare nel meccanismo di generazione del filopodio, la causa della riduzione di spine presente nel topo con delezione di MeCP2. È noto che tali meccanismi coinvolgono modificazioni dello scheletro interno della spina (il citoscheletro della spina) derivanti da modifiche biochimiche dell’actina, e che necessitano dell’attivazione della sintesi proteica a livello della spina dendritica stessa.
Guidati da queste considerazioni, abbiamo recentemente iniziato lo studio di questi due meccanismi molecolari nel nostro laboratorio. In particolare, per quanto riguarda la sintesi proteica, un precedente studio aveva mostrato che nel cervello di topi maschi con mutazione di MeCP2 era presente una ridotta attivazione della via molecolare mTor/AKT/S6 chinasi che risultava in una ridotta fosforilazione della proteina ribosomale S6 e una ridotta sintesi proteica. La ridotta fosforilazione di S6 era presente anche nelle femmine e, durante lo sviluppo, compariva intorno alle quattro settimane, ossia all’insorgere dei principali sintomi7.
È importante osservare che la via molecolare mTor/AKT/S6 chinasi ha tra i suoi attivatori l’IGF1, un fattore per cui è stata dimostrata un’azione migliorativa dei sintomi mostrata nel modello di delezione di MeCP2, sviluppato nel laboratorio di R. Janisch a Boston8. Abbiamo, quindi, deciso di utilizzare la tecnica di imaging al due fotoni per verificare se la somministrazione di IGF1 potesse stimolare la crescita di nuove spine dendritiche.
L’analisi ha mostrato che un’iniezione sistemica di IGF1 causa 24 ore dopo un aumento della produzione e della dinamicità dei filopodi6. Sebbene, il tasso di formazione dei filopodi presenti nei topi con mutazione di MeCP2 trattati con IGF1 non raggiungessero i valori dei topi normali, risultava comunque significativamente maggiore di quello dei topi con mutazione MeCP2 trattati con una soluzione salina di controllo.
Questi risultati rinforzano la possibilità che trattamenti che stimolino la sintesi proteica possano migliorare il fenotipo sinaptico dei topi con delezione di MeCP2. Inoltre, mostrano come l’imaging delle spine dendritiche al microscopio a 2 fotoni possa essere utilizzato per una valutazione relativamente veloce della efficacia di trattamenti volti a migliorare il fenotipo sinaptico ed i sintomi della sindrome di Rett.
BIBLIOGRAFIA
1 Derecki, N. C. et al. Wild-type microglia arrest pathology in a mouse model of Rett syndrome. Nature 484, 105-109, (2012).
2 Lioy, D. T. et al. A role for glia in the progression of Rett’s syndrome. Nature 475, 497-500, (2011).
3 Boggio, E. M., Lonetti, G., Pizzorusso, T. & Giustetto, M. Synaptic determinants of rett syndrome. Front Synaptic Neurosci 2, 28, (2010).
4 Armstrong, D. D. Neuropathology of Rett syndrome. Ment Retard Dev Disabil Res Rev 8, 72-76 (2002).
5 Holtmaat, A. & Svoboda, K. Experience-dependent structural synaptic plasticity in the mammalian brain. Nat Rev Neurosci 10, 647-658, (2009).
6 Landi, S. et al. The short-time structural plasticity of dendritic spines is altered in a model of Rett syndrome. Sci Rep 1, 45, (2011).
7 Ricciardi, S. et al. Reduced AKT/mTOR signaling and protein synthesis dysregulation in a Rett syndrome animal model. Hum Mol Genet 20, 1182-1196, (2011).
8 Tropea, D. et al. Partial reversal of Rett Syndrome-like symptoms in MeCP2 mutant mice. Proc Natl Acad Sci USA 106, 2029-2034, (2009).
