Negli ultimi anni il team di ricerca della Genetica Medica di Siena diretto dalla Professoressa Alessandra Renieri si è focalizzato sullo studio della terapia genica in diverse malattie, fra cui la Sindrome di Rett (RTT). In particolare, il team sta lavorando sulla correzione di mutazioni nei geni MECP2, FOXG1 e CDKL5, che causano rispettivamente la forma classica, la forma congenita e la variante con convulsioni a esordio precoce della malattia.
Per quanto riguarda il gene FOXG1, il team si è focalizzato sullo studio di due mutazioni: c.688C>T (p.(Arg230Cys)) e c.765G>A (p(Trp255Ter)). FOXG1 è un fattore di repressione della trascrizione, cioè spegne l’espressione di altri geni, e ha un ruolo essenziale per lo sviluppo del cervello; in particolare la corretta espressione di FOXG1 è fondamentale per il mantenimento della proliferazione dei progenitori neuronali, le cellule da cui deriveranno i neuroni. La correlazione tra FOXG1 e variante congenita della RTT è stata dimostrata per la prima volta nel 2008 grazie al lavoro del laboratorio della Genetica Medica che ha identificato le mutazioni sopra citate in due pazienti. Dato il ruolo fondamentale del gene FOXG1, risulta fondamentale correggerne la sequenza in modo preciso senza alterare ulteriormente la sua espressione.
Per tale motivo, il team di ricerca della Genetica Medica ha preso in considerazione l’impiego della terapia genica, un approccio terapeutico innovativo che permette di correggere una sequenza di DNA in modo preciso. La più comune forma di terapia genica è il gene replacement che prevede l’aggiunta di una copia addizionale del gene per sopperire alla funzione di quello mutato; questo processo risulta rischioso per quei geni implicati nella regolazione trascrizionale, come FOXG1, in quanto sono geni per i quali è essenziale non solo la presenza, ma anche il corretto dosaggio; farne produrre troppo ad una cellula, o nel momento e/o nella cellula sbagliata potrebbe inficiare ulteriormente sulla sua già alterata espressione. Per tale motivo, il gruppo della Prof.ssa Renieri ha scelto un approccio di terapia genica basato sul gene editing, che consente di correggere in modo preciso una specifica sequenza all’interno del gene di interesse senza alterarne i meccanismi di regolazione. L’obiettivo finale è quello di correggere la mutazione direttamente nei neuroni, il tipo cellulare primariamente coinvolto nella RTT, utilizzando la tecnologia CRISPR/ Cas9 e sfruttando uno dei meccanismi di riparazione della cellula, l’Homology-Directed Repair-HDR. Come modello per lo studio di questo approccio, sono state scelte le cellule staminali pluripotenti indotte o iPSC. Le iPSCs vengono ottenute per riprogrammazione a partire dai fibroblasti, ovvero cellule del tessuto connettivo che si ottengono tramite una semplice biopsia cutanea nel paziente; una volta ottenute possono essere indotte a diventare diversi tipi di cellule, tra cui i neuroni e permettono quindi di avere accesso a cellule derivate dal paziente e che sono quelle primariamente interessate dalla malattia.
Per la correzione delle mutazioni, in collaborazione con l’Istituto Toscano Tumori (ITT), sono stati disegnati e ingegnerizzati due plasmidi (sequenze di DNA circolare normalmente presenti all’interno delle cellule batteriche) che esprimono i componenti fondamentali per il gene editing. Il macchinario molecolare che corregge le mutazioni ha come attore principale una proteina chiamata Cas9 che funziona come una forbice molecolare, ovvero taglia il DNA nel sito specifico della mutazione e innesca i meccanismi di riparazione e il successivo ripristino della sequenza corretta. La scelta di lavorare con due plasmidi nasce dall’esigenza di utilizzare i virus Adeno-Associati (Adeno Associated Virus – AAVs) come sistema per far arrivare all’interno delle cellule, in vitro e in vivo, i componenti del macchinario di editing. Gli AAVs sono dei virus che non possono contenere RNA di dimensioni elevate, ma rappresentano un sistema virale più sicuro rispetto ad altri sistemi virali utilizzati in terapia genica. Nel nostro caso è stato scelto il sierotipo AAV9 che è in grado di raggiungere il sistema nervoso centrale attraversando la barriera emato-encefalica ed infetta preferenzialmente le cellule neuronali. Il sistema basato sull’utilizzo di due plasmidi è stato recentemente brevettato dall’Università di Siena.
I primi test di editing su cellule con le due mutazioni sopra-citate nel gene FOXG1 hanno dato risultati estremamente promettenti. In primo luogo, sono stati trasfettati i fibroblasti dei pazienti per valutare rapidamente la funzionalità del sistema; l’attivazione e l’efficienza dei plasmidi sono state validate qualitativamente e quantitativamente utilizzando il microscopio a fluorescenza e la citometria a flusso (Fluorescence-Activated Cell Sorting – FACS). Il FACS, grazie ad un sistema fluorescente, permette di selezionare e recuperare solo le cellule potenzialmente corrette. La conferma del ripristino della sequenza corretta avviene grazie al sequenziamento di nuova generazione (Next Generation Sequencing – NGS). L’efficienza di correzione, valutata tramite NGS, nei fibroblasti con entrambe le mutazioni testate è di circa il 22% e risulta paragonabile in test indipendenti. Inoltre il sistema di correzione è stato validato nei neuroni con la mutazione c.688C>T (p.(Arg230Cys)), ottenendo un’efficienza del 34%. Per iniziare a valutare se la correzione della mutazione risulti nella normalizzazione di altri parametri della cellula alterati dalla ridotta funzionalità di FOXG1, sono stati analizzati nei precursori neuronali i livelli di espressione della proteina PAX6. PAX6, insieme a FOXG1, ha un ruolo importante nella proliferazione dei progenitori neuronali e risulta espresso in eccesso nei precursori neuronali che presentano la mutazione in FOXG1. Le analisi hanno dimostrato che, in seguito al trattamento di queste cellule con il sistema di editing, si ha il ripristino dei normali livelli di questa proteina confermando l’effetto positivo della correzione. I risultati di questo studio sono stati recentemente pubblicati sullo European Journal of Human Genetics.
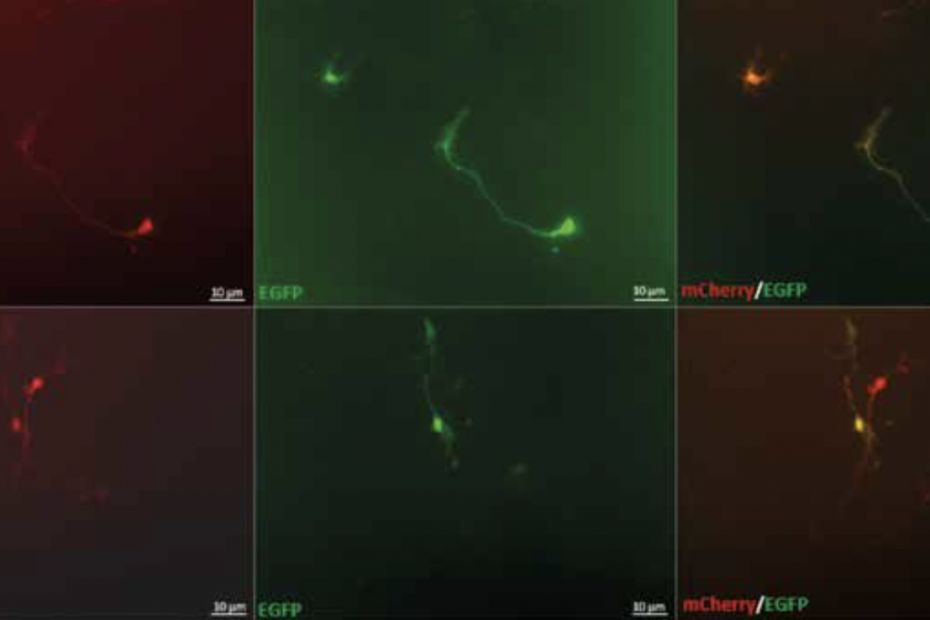
Questi risultati dimostrano che è possibile fare gene editing nelle cellule del sistema nervoso utilizzando l’HDR e rappresentano un buon punto di partenza per i prossimi esperimenti di editing che prevedono di attuare la stessa strategia utilizzando i virus. Esperimenti preliminari in vitro hanno dimostrato che i neuroni infettati con AAV9 che codificano le componenti del sistema di correzione mutazionespecifico esprimono correttamente il sistema (Fig.1).
Lo step successivo sarà quello di validare l’efficienza di HDR in vivo in topi knock-in, cioè animali nei quali è stata inserita la stessa mutazione presente nei pazienti.