
Sunto dell’articolo scientifico “Radically truncated MeCP2 rescues Rett syndrome-like neurological defects”. Autori: Tillotson R, Selfridge J, Koerner MV, Gadalla KKE, Guy J, De Sousa D, Hector RD, Cobb SR, Bird A., Pubblicato su: Nature. 2017 Oct 19;550(7676):398-401.
a cura di Maurizio d’Esposito e Silvia Russo
Il panorama sul trattamento della Sindrome di Rett è radicalmente mutato da quando, nel 2007, il gruppo di Adrian Bird ha dimostrato che la malattia può essere curata facendo esprimere un gene MECP2 funzionante in cellule di topo malate, ovvero che non erano più in grado di produrre la proteina MECP2. Lo studio aveva evidenziato che questo tipo di intervento è efficace anche nel periodo postnatale, cioè quando il quadro sintomatologico della patologia è già evidente (Guy et al., 2007).
Questo ha portato all’attuazione di progetti di terapia genica, che mirano a reintrodurre il gene MECP2 integro (funzionante) sostituendolo al gene mutato (malfunzionante) in tutte le cellule del corpo, ma in particolar modo nel cervello, organo bersaglio della malattia.
La terapia genica è una tecnologia molto promettente, ma è influenzata da diversi parametri:
- L’utilizzo di molecole trasportatrici (vettori) che riescano ad impacchettare il gene integro, con le sue regioni regolative principali. I vettori principalmente utilizzati sono gli adenovirus, che però hanno una capacità limitata di integrare nel loro DNA le sequenze utili del gene in questione.
- La capacità di trasdurre (cioè di far arrivare all’organo bersaglio, nel nostro caso il cervello) il vettore adenovirale con il suo gene integro inserito. Se infettiamo con poche molecole di adenovirus, il risultato è nullo; se utilizziamo troppe molecole di adenovirus, abbiamo gravi effetti collaterali, dovuti a tossicità (Gadalla et al., 2017).
- La somministrazione è fino ad ora sistemica, cioè la molecola si diffonde in tutto il corpo in seguito ad iniezione nel sangue. Negli animali l’iniezione intracerebrale aumenta notevolmente l’efficienza di trasduzione, ma questa modalità di somministrazione non può essere praticata nell’uomo.
Il lavoro del gruppo di Adrian Bird, pubblicato il 19 ottobre 2017 (Tillotson et al., 2017) apre una nuova possibilità per l’utilizzo di vettori adenovirali che possano trasdurre efficacemente un gene MECP2 funzionante nelle cellule malate. Infatti, Tillotson e collaboratori hanno dimostrato che una proteina MECP2 notevolmente più piccola del normale è capace di sanare quasi totalmente le manifestazioni patologiche (fenotipo) in un modello di topo della Sindrome di Rett.
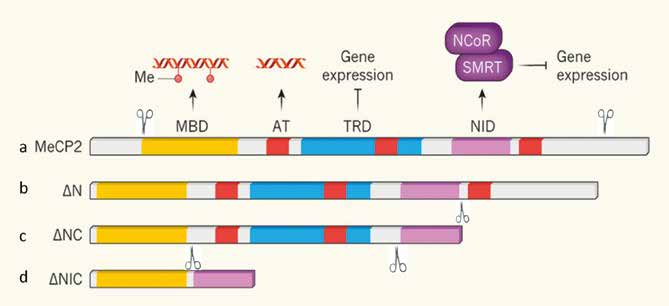
Fig. 1 (adattata dall’articolo di AE West, 2017). a) lo schema rappresenta la proteina MeCP2 con tutti i suoi domini funzionali: MBD, AT, TRD e NID; b e c) rappresentano due versioni, ΔN e ΔNC, della proteina in cui è stata rimossa la porzione iniziale e terminale rendendola più piccola, ma senza togliere i domini funzionali più importanti; d) in quest’ultimo schema è mostrata la versione più piccola della proteina costruita nel laboratorio di Bird; ΔNIC, a cui sono stati rimossi i domini AT e TRD. I tre costrutti come spiegato nel testo non conferiscono un quadro clinico patologico (casi b e c) o solo lievemente, caso d.
La perdita di regioni della parte iniziale della proteina (NH-terminale) e della parte finale della stessa (COOH-terminale) nell’animale transgenico (portatore della proteina difettosa) non comporta conseguenze cliniche né effetti molecolari (il legame con il DNA e con altre proteine resta invariato).
Nella sua accezione più estrema, è sufficiente una proteina MECP2 lunga 1/3 della proteina normale, e che contenga esclusivamente il sito di legame con il DNA metilato (MBD) ed il sito di legame a co-repressori (NCor-SMRT) per migliorare efficacemente parte dei sintomi neurologici della patologia RTT in topo.
Questa scoperta evidenzia la possibilità di utilizzare in modo efficace vettori adenovirali capaci di avere nel loro interno tutte le regioni regolative necessarie alla corretta espressione del gene MECP2 nelle cellule malate.
Sebbene si tratti di scoperte incoraggianti per la cura della RTT, bisogna precisare che il passaggio dal modello sperimentale murino al trattamento delle pazienti non è immediato.
Restano infatti alcuni limiti tecnici che gli scienziati cercano alacremente di superare. Per esempio, al fine di ridurre al minimo gli effetti collaterali legati all’uso di virus, bisognerà trovare le strategie opportune per iniettare la minore quantità possibile di adenovirus direttamente nelle cellule malate. Dunque, oltre a trovare un’alternativa alla somministrazione sistemica del vettore adenovirale, occorre disegnare/individuare nuovi vettori che possano attraversare con efficienza la barriera emato-encefalica e che, magari, siano capaci di raggiungere specificamente le cellule malate.
La sfida è ancora aperta: ora che sappiamo qual è la porzione minima di Mecp2 utile alla terapia genica bisognerà concentrarsi su come rendere efficiente ed efficace il “rimpiazzo” del gene malato con il gene sano.
Bibliografia essenziale
- Gadalla KKE, Vudhironarit T, Hector RD, Sinnett S, Bahey NG, Bailey MES, Gray SJ, Cobb SR. 2017. Development of a Novel AAV Gene Therapy Cassette with Improved Safety Features and Efficacy in a Mouse Model of Rett Syndrome. Mol Ther Methods Clin Dev 5:180-190.
- Guy J, Gan J, Selfridge J, Cobb S, Bird A. 2007. Reversal of neurological defects in a mouse model of Rett syndrome. Science 315:1143-1147.
- Tillotson R, Selfridge J, Koerner MV, Gadalla KKE, Guy J, De Sousa D, Hector RD, Cobb SR, Bird A. 2017. Radically truncated MeCP2 rescues Rett syndrome-like neurological defects. Nature 550:398-401.
